L'ostéogenèse imparfaite

- Détails
- Mis à jour le 24 juin 2012
L’ostéogenèse imparfaite (O.I.) est une affection héréditaire caractérisée par une fragilité osseuse avec des fractures multiples pour des traumatismes minimes.
C’est une maladie rare (1 cas /15 à 20 000 naissances) sans facteur ethnique ni racial particulier [1-2-3]. Elle est due à une anomalie de production du collagène de type I qui peut être qualitative ou quantitative. Il en résulte un groupe hétérogène de maladies allant de la forme létale à la naissance aux formes bénignes et ou découvertes à l’age adulte. L’avènement des bisphosphonates et de l’enclouage télescopique en a modifié le pronostic notamment chez l’enfant.
PHYSIOPATHOLOGIE
Le collagène de type I constitue 90% de la matrice organique de l’os mais aussi des tissus conjonctifs : tendons derme dentine… Dans l’O.I. la minéralisation (dépôt d’apatite) se trouve réduite car la trame collagène qui constitue le lit est altérée en nombre ou en qualité. L’os formé est donc fragile.
En microscopie les molécules de collagène sont formées par 3 chaînes polypeptidiques ; 2 chaînes identiques α1(I) codées par le gène COL1A1 situé sur le chromosome 17 et une chaîne α2(I) codée par le gène COL1A2 situé sur le chromosome 7 [4]. Les 3 chaînes constituent une triple hélice enroulée dont le centre ne peut être occupé que par la glycine : le plus petit acide aminé (AA). Toute substitution par un autre AA (mutation) entraînera une déformation importante avec ralentissement de la formation des fibres de collagène. La sévérité dépend de la position de la mutation (plus elle est proche du début de la chaîne plus elle est sévère) [5] l’atteinte de α2 (I) étant plus sévère que celle de α1 (I). La nature de l’AA substitué a aussi un rôle pronostique (l’arginine est plus sévère que la cystéine). Tout ceci a pour corollaire une multitude de mutations et des phénotypes très variés d’O.I.
CLASSIFICATION
L’objectif majeur d’une classification est d’établir la relation entre le génotype et les manifestations cliniques qui en résultent. Le problème n’est cependant pas simple devant la multitude des mutations et le fait que le collagène type III peut se substituer au collagène I ce qui explique que d’autres tissus (peau tendons ligaments…) sont peu ou pas touchés. L’atteinte de la dentine reste elle sans réponse.
La classification de SILLENCE en 4 types est la plus utilisée. Actuellement en réponse à des formes intermédiaires la classification a été étendue à 7 groupes par GLORIEUX (tableau I) [6].
Tableau I : Classification de l’ostéogenèse imparfaite (O.I.) de Sillence et de Glorieux.[6]
|
1. O.I. de type I (bénigne) |
Fractures par suite de traumatismes minimes Sclérotique bleutée Malformation minime des os longs Taille normale ou quasi-normale Possibilité de dentinogenèse imparfaite |
|
2. O.I. de type II (mortelle) |
Fractures intra-utérines Chapelet costal Sclérotique bleutée Fémur large et court Détresse respiratoire Décès pendant la période périnatale |
|
3. O.I. de type III (grave) |
Fractures fréquentes par suite de traumatismes minimes Sclérotique de couleur variable Taille extrêmement petite Grave malformation des membres Scoliose Faciès triangulaire Dentinogenèse imparfaite fréquente |
|
4. O.I. de type IV (modérée) |
Fractures par suite de traumatismes minimes Sclérotique de couleur variable Taille modérément petite Malformation modérée des membres Scoliose Possibilité de dentinogenèse imparfaite |
|
5. O.I. de type V |
Fractures par suite de traumatismes minimes Sclérotique normale* Calcification de la membrane interosseuse de l’avant-bras ou de la jambe Bande métaphysaire dense sous la plaque de croissance Collagenèse hypertrophique par suite de fractures ou de bâtonnets intramédullaires Absence de dentinogenèse imparfaite |
|
6. O.I. de type VI |
Fractures par suite de traumatismes bénins Sclérotique normale* élévation modérée du taux de phosphatase alcaline Stries de Looser (pseudofractures) visibles à la radiographie Absence de dentinogenèse imparfaite Absence d’os wormiens Plus : Absence de rachitisme |
|
7. O.I. de type VII |
Fractures par suite de traumatismes bénins Sclérotique normale * Absence de dentinogenèse imparfaite Coxa vara Rhizomélie (brièveté des racines des membres supérieurs et inférieurs) |
*Sclérotique normale = blanche ou légèrement bleutée
O.I. ET TRANSMISSION GENETIQUE
Quand l’un des parents est porteur de l’allèle pathologique la transmission est de type autosomique dominante. La probabilité de transmettre l’affection est de l’ordre de 50%. Des cas sporadiques d’enfants malades issus de parents non affectés ont été décrits. Des cas de transmission récessive existent aussi ce qui rend le conseil génétique parfois difficile.
Le diagnostic prénatal constitue actuellement une avancée et peut être posé dès la 18ème semaine de gestation [7] grâce à l’échographie 3D et le scanner hélicoïdal [89]. Récemment l’IRM a été introduite. Elle permet de confirmer le diagnostic avec plus de précision [10]. Une anomalie de la transparence osseuse au cours des examens de suivi d’une grossesse doit attirer l’attention.
La grossesse chez les femmes atteintes d’O.I. se passe en général sans problème mais des incidents à type de douleurs atroces lombaires avec des fractures du bassin et des vertèbres ont été décrites avec une fréquence avoisinant 13% [11]. Des cas d’accouchement par césarienne ont été pratiqués avec un bon pronostic fonctionnel et vital pour le nouveau né.
LES MANIFESTATIONS CLINIQUES
Indépendamment de la sévérité l’O.I. associe certains signes cliniques :
-l’ostéoporose : elle constitue un problème majeur car elle est responsable de la fragilité osseuse. La densité minérale osseuse (DMO) n’est pas corrélée à la classification de SILLENCE [12] mais serait en rapport avec le poids corporel des patients. L’ostéodensitométrie constitue la technique de choix pour le diagnostic. Anodine et répétitive elle permet d’analyser le contenu minéral osseux sur un os de faible quantité minérale [13].
-les fractures: sont souvent le premier signe d’appel. Elles surviennent après des traumatismes minimes. Elles intéressent les os plats (côtesvertèbres) mais surtout les diaphyses des os longs la consolidation se fait dans les mêmes délais qu’un os normal avec une fréquence accrue de pseudarthrose (Fig. 1) [14] les cals vicieux peuvent également se voir aussi bien chez les femmes que chez les hommes. Le diagnostic de ces cals doit se faire avec circonspection car des cas d’ostéosarcome peuvent prêter le change [15]. Dans le type II elles surviennent tôt dès la naissance et engagent le pronostic vital. Dans les formes modérées leur incidence augmente avec les premiers pas et en péripubérté .Chez les filles du fait de l’imprégnation oestrogénique L’incidence des fractures diminue de l’âge de la puberté à la ménopause.
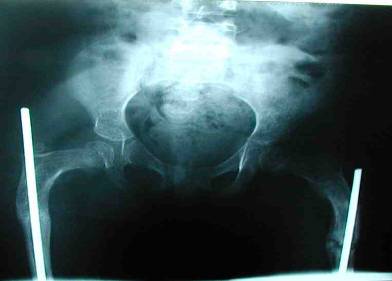
Fig. 1 : Fractures diaphysaires d’age différent chez une fillette de 6 ans.
-les déformations osseuses : peuvent être secondaires à des cals vicieux sur fracture avec angulation. Elles restent une source de faiblesse mécanique.
Parfois ces déformations sont spontanées car l’os fragile en grandissant n’arrive pas à étirer les muscles et les parties molles adjacentes il s’en suit plusieurs aspects :
aspect en crosse antérieure ou antéro-externe du fémur. aspect en lames de sabre au tibia (Fig. 2). aspect de fausse coxa vara au niveau des hanches car elle est réductible. des protrusions acétabulaires. le thorax peut être le siège d’un pectus excavatum ou carinum avec réduction des capacités respiratoires.
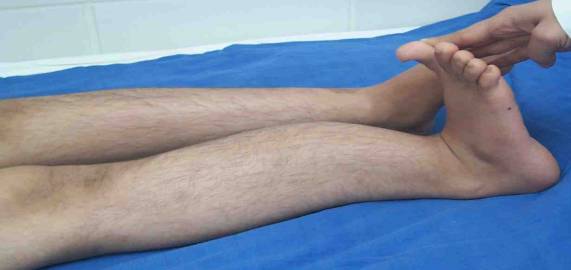
Fig. 2 : déformation du tibia associée à une hyper laxité ligamentaire.
-les déformations du rachis : méritent un intérêt particulier de part leur fréquence de 20 à 80% tous types d’O.I. confondus [16]. L’hyperlaxité ligamentaire et la fragilité osseuse en sont les plus grands pourvoyeurs. La scoliose reste très fréquente et pose un problème de prise en charge. Son incidence varie de 30 à 100% selon les formes d’O.I. et pour un angle de Cobb supérieur ou égal à 30° il existe une corrélation significative avec la baisse de la densité osseuse [17].
D’autres déformations peuvent se voir avec notamment au niveau du crâne une macrocéphalie et la présence d’os wormiens à la radiologie.
-autres manifestations :
l’atteinte des sclérotiques : la coloration bleue est l’apanage du type I (Fig. 4) elle est noire dans le type II et peut être grise dans le type III et IV. Le collagène anormal augmente la transparence de la cornée.
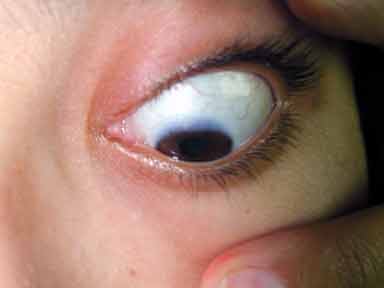
Fig. 4 : Aspect bleuté des sclérotiques dans un type I.
la surdité : fréquente elle peut être de transmission (fracture de l’étrier) ou de perception (atteinte de la capsule optique). Il n’y a pas de corrélation entre le type de mutation COL1A1 ou A2 et la survenue de la surdité [18]. Actuellement il est établi qu’elle n’est plus l’apanage des patients de la 20 à 30 aine et sa fréquence est élevée chez l’enfant pouvant atteindre 773 % à l’âge de 9 ans [19]. Une surveillance par audiogramme est donc indispensable et le traitement varie de l’appareillage à la stapedectomie. l’hyperlaxité ligamentaire : se voit chez 70% des patients [20]. Le pied plat en est la manifestation la plus fréquente. L’hyperlaxité est en général bien tolérée sauf chez les sujets très actifs [21]. la dentinogenèse imparfaite : elle intéresse surtout les dents de lait et se voit dans 25% des cas [22]. Les dents sont rapidement usées les canaux pulpaires oblitérés l’email fragile d’où la nécessité d’une hygiène régulière avec des fluorés. Parfois des couronnes et même des implants sont nécessaires. autres :
+ L’impression basilaire est une complication neurologique qu’il convient d’évoquer devant des céphalées avec hyper réflexie et atteinte des dernières paires crâniennes. Actuellement grâce à l’IRM le diagnostic est fait tôt.
+ L’atteinte cardio-pulmonaire peut mettre en jeu le pronostic vital. Elle peut être à type d’insuffisance aortique ou mitrale ou d’insuffisance respiratoire avec une fréquence accrue des pneumonies.
+ La fragilité capillaire est fréquente au cours de l’O.I. [23] comme en témoigne la présence spontanée d’hématomes.
DIAGNOSTIC DIFFERENTIEL
L’O.I. peut faire évoquer certaines pathologies notamment :
-une hypophosphatasie infantile qui se manifeste par une ostéoporose sévère avec micromilie et un taux bas de phosphatases alcalines.
-une ostéoporose juvénile primaire ou secondaire soit à un hypogonadisme soit à une pathologie maligne.
-le véritable problème qui se pose est celui des enfants battus ou syndrome de Silverman d’où la nécessité de mesurer la densité minérale osseuse et rechercher des signes physiques de maltraitance.
TRAITEMENT
Ne se conçoit qu’avec une approche multidisciplinaire.
A. Traitement médical :
Plusieurs thérapeutiques ont été essayées (fluor calcitonine oestrogènes androgènes thyroxine….) sans qu’aucune ne fasse preuve d’efficacité. En fait le traitement médical a connu ses titres de noblesse avec l’avènement des bisphosphonates et depuis les essais ne cessent de se multiplier. Le premier mérite revient à Devogelaer et al. qui ont noté en 1987 une amélioration clinique et radiologique chez un garçon de 12 ans atteint d’O.I. [24].
Le bisphosphonate le plus utilisé est le pamidronate par voie parentérale mais des formes orales telles :l’olpadronate (médicament en cours d’essai)[25] ; l’alendronate donné à 5 ou 10 mg par jour en fonction du poids ; et le risédronate ont également fait preuve de leur efficacité.
Plusieurs effets bénéfiques ont été notés sous bisphosphonates :
Sur le plan clinique :
+ on note une nette baisse des douleurs osseuses quelques semaines après la 1ère injection [2627].
+ une sensation de bien être [28].
+ une augmentation de la force musculaire [29].
+ une augmentation annuelle très nette de la taille et du poids.
++ une diminution du nombre de fractures parfois de l’ordre de 65% et ceci pendant et après traitement car l’effet des bisphosphonates persiste plusieurs années dans le tissu osseux [30].
Sur la DMO
ü toutes les études convergent vers un gain de la masse osseuse [31-32]. En outre les vertèbres tassées reprennent leur taille initiale.
ü le gain de la DMO peut atteindre 25% mais ceci doit être interprété avec prudence chez l’enfant car il est en pleine croissance.
· Sur les marqueurs de remodelage osseux :
Glorieux trouve une nette diminution annuelle des phosphatases alcalines de l’ordre de 13% et une baisse plus conséquente des crosslaps urinaires (26%) [33].
· Histomorphométrie :
Le traitement par bisphosphonates a montré une augmentation considérable de l’épaisseur de la corticale ce qui rend la rééducation facile et la chirurgie plus aisée sur un os solide.
Modalités de prescription : pour le Pamidronate.
Chez l’adulte la dose est en général de 90 mg une fois tous les 3 mois. Chez l’enfant les modalités peuvent être résumées comme suit :
Moins de 2 ans : 05 mg/kg/j x 3 jours tous les 2 mois. 2 à 3 ans : 075 mg/kg/j x 3 jours tous les 3mois Plus de 3 ans : 1mg/kg/j x 3 jours (max 60mg) tous les 4 mois
Actuellement le traitement doit débuter tôt chez les enfants et surtout dans les 2 premières années de la vie [34].
Lors de la première injection une surveillance attentive des enfants s’avère indispensable car en plus des effets connus des bisphosphonates (fièvre syndrome grippal…) de rares cas de détresse respiratoire ont été signalés.
La supplémentation vitamino-calcique :
Si le dosage de la calcémie est souvent normal une hypercalciurie est parfois retrouvée. De même le dosage de la 25 OH vitamine D est bas [3536] ce qui justifie l’instauration d’un traitement à base de calcium et de vitamine D chez les patients atteints d’O.I.
O.I. et parathormone (tériparatide ou PTH) :
Par extrapolation au traitement de l’ostéoporose post-ménopausique certains essais thérapeutiques à base de PTH ont été réalisés chez les rats [37]. Mais pour le moment ce traitement n’a pas de place dans l’O.I.
O.I. et hormone de croissance (GH) :
L’étude anthropométrique des enfants atteints d’O.I. a montré une nette réduction de leur taille et un taux effondré de l’IGF-I et IGFBP3 [38]. Après un traitement à base de GH (dose de 01 à 02 m/kg/j 6 jours/semaine) les enfants augmentent de 50% leur vitesse de croissance. Un gain DMO est noté aussi. Comme facteur prédictif à une bonne réponse au traitement il faut signaler l’existence d’un taux élevé du carboxyterminal peptide [39].
L’association GH et bisphosphonates dans le traitement de l’O.I. semble très prometteuse mais pour le moment les essais sont disparates et ne concernent que des petites cohortes.
O.I. et greffe de moelle :
Des essais cliniques ont été entrepris mais sans grande conviction [40-41].
B. Le traitement chirurgical :
Le risque anesthésique n’est pas négligeable (fractures intubation difficile détresse respiratoire .. .) d’où la nécessité d’une équipe entraînée.
-os longs : Les plaques vissées sont contre indiquées en cas d’O.I. (risque de fracture…). Les clous télescopiques sont les plus utilisés. Leur petite taille (de 32 mm à 64 mm) permet une adaptation pour chaque os. Ils peuvent être introduits par voie transcutanée avec respect du cartilage de conjugaison. Leur propriété d’allongement n’altère pas la croissance. Une fois celle-ci achevée ils peuvent être remplacés par des clous classiques. L’enclouage permet une nette diminution du nombre de fractures une correction des déformations et une reprise de l’autonomie en effet 75% des enfants reprennent la marche contre 40% avant l’intervention [42]. Pour les types III et IV on conseille l’intervention dès l’âge de la marche.
-rachis : le corset est de maniement difficile en cas d’O.I. (moulage difficile sur un thorax mou complications respiratoires…). La chirurgie notamment l’arthrodèse vertébrale postérieure reste le traitement de choix elle stabilise et permet la position assise chez le patient en fauteuil roulant.
-Quand intervenir ?
Si l’angle de Cobb est moins de 20° on se contente d’une expectative. Pour le type I et IV l’arthrodèse est indiquée pour un angle supérieur ou égal à 45°. Pour le type III très déformant l’arthrodèse est indiquée pour un angle supérieur ou égal à 30°.
C. La prise en charge rééducative :
Elle constitue la charpente de la prise en charge des patients atteints d’O.I. Elle indiquée dès la naissance en apprenant aux parents à manipuler leur enfant sans très grande appréhension. Elle lutte également contre la douleur et la perte de l’autonomie en jouant un rôle positif sur la proprioception et la trophicité osseuse et musculaire. Elle vise aussi le maniement du fauteuil roulant avec transfert des forces aux membres supérieurs. Elle aide également à la verticalisation et à la reprise de la marche. La balnéothérapie en plus de ses vertus myorelaxantes et antalgiques permet un travail respiratoire et contribue au développement des muscles para rachidiens.
D. O.I. et thérapie génique :
Constitue à elle le vrai traitement étiologique. C’est une voie très convoitée par beaucoup d’équipes. Le principe est de remplacer ou d’atténuer le gène défectueux mais elle se heurte à de nombreux obstacles. Dans certains travaux utilisant des ribosomes transfectés par des virus près de 50% du RNAm du collagène muté a été éliminé. Ceci reste un premier pas pour un long chemin [43].
E. Aspect psycho-social :
Longtemps isolés les parents de patients atteints d’O.I. peuvent désormais adhérer à des associations d’O.I. où ils peuvent partager leurs soucis mais aussi leurs expériences.
CONCLUSION
L’ostéogenèse imparfaite reste une maladie rare hétérogène par sa présentation. Certes beaucoup de progrès ont été acquis pour sa prise en charge qui doit rester toujours multidisciplinaire en attendant une future thérapie génique opérante.
Van der Rest M. Biologie du collagène et maladies héréditaires de la matrice extracellulaire. Médecine/sciences 1987;3:411-20 Sillence DO Senn A Danks DM.Genetic heterogenety in O.I .J med genet 1979;16:101-16. Van der Rest M. Garrone R. Collagen family of proteins. In FASEB J. 1991; 5:2814-23 Marini JC Letocha AD. Osteogenesis imperfecta. Endotext.com-diabetes july22 2002 1-12. Rauch F Glorieux FH. Osteogenesis imperfecta. Lancet 2004; 363: 1377-85 Glorieux FH Rauch F Plotkin H et al. Type v O.I : a new form of brittle bone disease.J Bone Miner Res 2000;15(9) : 1650-8. Fassier F. Ostéogenèse imparfaite de l’enfant. Cahiers d’enseignement de la SOFCOT 1999 : 235-52 Dhouib M Guirat N. Lethal osteogenesis imperfecta. Prenatal diagnosis. Presse Med 2004 ; 33 : 658-60 Ruano R Molho M Roume J Ville Y. Prenatal diagnosis of fetal skeletal dysplasias by combining two-dimensional and three-dimensional ultrasound and intrauterine three-dimensional helicoidal computer tomography. Ultrasound Obstetr Gynecol 2004; 24 : 134-40 Teng SW Guo WY Shen MH Wang PH. Initial experience using magnetic resonance imaging in prenatal diagnosis of osteogenesis imperfecta. Calcif Tissue Int 2003;73: 441-5 Glorieux FH Lanone G Chabot G Travers R. Bone mineral density in O.I; J Bone Min Res 1994;9s1[abstract]:s225. McAllion SJ Paterson CR. Musculo-skeletal problems associated with pregnancy in women with osteogenesis imperfecta. J Obstet Gynaecol 2002; 22: 169-72 Kok DJ Viterwaal CS Van Dougen AJ et al. The interaction between Sillence type and BMD in osteogenesis imperfecta. Calcif Tissue Int 2003;73 : 441-5 Gamble JG Rinsky LA StrudwickJ Bleck EE. Non-union of fractures in children who have osteogenesis imperfecta. J Bone Joint Surg 1988; 70A: 439-43 Takahashi S Okada K Nagasawa H Schimada Y Sakamato H. Osteosarcoma occuring in osteogenesis imperfecta. Virchows Arch 2004; 444 : 454-8 Lubicky JP. The spine in osteogenesis imperfecta. In: Weinstein SL. The pediatric spine: principles and practice : 943-58. New York Raven Press Ltd;1994. Papin P Fassier F Hamdy R Glorieux FH Duhaime M. Can DEXA and Sillence classification predict the development of scoliosis in patients with osteogenesis imperfecta? SICOT Poster n°0138.Sidney Australia 1999. Hartikka H Kuurila K Korkko J et al. Lack of correlation between the type of COL1A1 or COL1A2 mutation and hearing loss in osteogenesis imperfecta patients. Hum Mutat 2004;24 :147-54 Imani P Vijayasekaran S Lannigan F. Is it necessary to screen for hearing loss in the pediatric population with osteogenesis imperfecta. Clin Otolaryngol 2003; 28 :199-202 Beighton PH Spranger J Versveld G. Skeletal complications in osteogenesis imperfecta. A review of 153 south african patients. South Afr Med J 1983; 64:565-8 Ogilivie-Harris DJ Khazim R. Tendon and ligament injuries in adults with osteogenesis imperfecta. J Bone Surg 1995; 77B: 155-6. Benichou O Kuntz D. L’ostéogenèse en 2000.In MF KahnD KuntzO MeyerT BardinP Orceled L’actualité rhumatologique 2000 ; Elsevier: 249-60. Evensen SA Myhre L Stormorken H. Haemostatic studies in osteogenesis imperfecta. Scand J Haematol 1984 ; 33 : 177-9 Devogelaer JP Malghem J Maldague B Naguant de deuxchaisnes C. Radiological manifestations of bisphosphonates treatment with APD in a child suffering from osteogenesis imperfecta. Skeletal Radiol 1987; 16 : 360-63 Sakkers R Kok D Engelbert R et al.Skeletal effects and fuctional outcome with olpadronate in children with osteogenesis imperfecta a 2 year randomised placebo-controlled study.Lancet.2004;363:1427-31 Huaux JP Lokietek W. Is APD a promising drug in the treatment of severe osteogenesis imperfecta? J Pediatr Orthop 1988; 8: 71-72 Glorieux FH Bishop NJ Plotkin H Chabot G Lanoue G Travers R. Cyclic administration of pamidronate in children with severe O.I. N eng J Med 1998; 339:947-52. Zacharin M. Bateman J. Pamidronate treatment of osteogenesis imperfecta. Lack of correlation between clinical severity age at onset of treatment response. J Pediatr Endocrinol Metab 2002; 15:163-74 Astrom E Soderhall S. Beneficial effect of longterm intravenous bisphosphonate treatment of osteogenesis imperfecta. Arch Dis Child 2002; 86:356-64 Montpetit K Plotkin H Rauch F et al. Rapid increase in grip force after start of pamidronate therapy in children and adolescents with severe osteogenesis imperfecta. Pediatrics 2003;11: 601-3 Garbner B Landis WJ Roscher P et al. Age- and genotype-dependance of bone material properties in the osteogenesis imperfecta murine model (oim). Bone 2001; 29:453-7 Benson DR Newman DC. The spine and surgical treatment in osteogenesis imperfecta. Clin Orthop 1981;159:147-53 Plotkin H Rauch F Bishop NJ et al. Pamidronate treatment of severe osteogenesis imperfecta in children under 3 years of age. J Clin Endocrinol Metab 2000; 85:1846-50 Kuivaniemi H Tromp G Prockop D. Mutations in fibrillar collagens (type IIIIIIXI) fibril-associated collagen (type IX) and network forming collagen (type X) cause a spectrum of diseases of bone cartilage and blood vessels. Human Mutat 1997; 9: 300-15 Chines A Petersen D Schranck F Whyte M. Hypercalciuria in children severely affected with osteogenesis imperfecta. J Pediatr 1991;119: 51-7 Vahle JL Sato M Long GG et al. Skeletal changes in rats given daily subcutaneous injections of recombinant human parathyroid hormone (1-34) for 2 years and relevance to human safety. Toxicol Pathol 2002;30: 312-21 Lund AM Muller J Skovby F. Anthropometry of patients with osteogenesis imperfecta. Arch Dis Child 1999;80: 524-8 Marini JC Hopkins E Glorieux FH Chrousos GP Reynolds J Gunberg CM Reing CM. Positive linear growth and bone responses to growth hormone treatment in children with types III and IV osteogenesis imperfecta high predective value of the carboxyterminal peptide of type 1 procollagen. J Bone Miner Res 2003; 18 :237-43 Byers PH. Osteogenesis imperfecta: perspectives and opportunities. Curr Opin Pediatr 2000;12: 603-09 Bianco P Gehron Robey P. Marrow stromal stem cells. J Clin Invest 2000;105:1663-68 Smith R. Severe osteogenesis imperfecta: new therapeutic options? BMJ 2001; 322: 63-4 Byers PH. Killing the messenger: new insights into nonsense-mediated m RNA decay. J Clin Invest 2002; 109: 3-6 Mollington-Ward S Allers CTuohy G et al. Validation in mesenchymal progenitor cells of a mutation-independant ex vivo approch to gene therapy for osteogenesis imperfecta. Hum Mol Genet 2002; 418:331-5

Articles les plus lus
- L'arthrose cervicale
- L'arthrose lombaire
- La spondylarthrite ankylosante
- Coxarthrose
- La fibromyalgie
- Les anti-inflammatoires non stéroïdiens. Modalités de prescription.
- Apport de l’échographie dans le diagnostic d’une épaule douloureuse.
- La gonarthrose
- Diagnostic précoce de la polyarthrite rhumatoïde
- La maladie de Behçet